|
 |
 |
 |
|
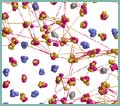
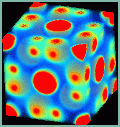
ProjectsAtomistic SimulationsThe focus of this project is on atomistic simulations. Classical Molecular Dynamics is used to simulate the dynamics of large systems, whereas abinitio simulations are used to gain a deeper understanding of the interactions between the atoms. IMD - ITAP Molecular DynamicsIMD is a software package for classical molecular dynamics simulations. It was developed at the Institut für Theoretische und Angewandte Physik (ITAP) in the framework of the Sonderforschungsbereich 382 „Verfahren und Algorithmen zur Simulation physikalischer Prozesse auf Hochleistungsrechnern“. Several types of interactions are supported, among them central pair potentials, EAM potentials for metals and Tersoff potentials for covalent systems. A rich choice of simulation options is available: different integrators simulating the various thermodynamic ensembles, relaxation integrators, force boundary conditions, facilities to shear or deform the sample during simulation, and many more. The software is flexible, modular and portable and achieves high performance on contemporary computer architectures. IMD runs efficiently on both single processor workstations and massively parallel supercomputers. The parallelization uses the standard MPI library for message passing. Applications:
ContactKatharina Benkert |
|